Zika Virus (ZIKV) is a mosquitoe-borne Flavivirus (MBFV), related to other Flavivirus’ that are transmitted by mosquitoes such as Dengue Virus (DENV), Yellow Fever Virus (YFV) or Tick Borne Encephalitis Virus (TBEV) and as such the viral genome is released into the cytoplasm following viral entry and translated into a single polyprotein that is cleaved by both viral and cellular proteases into the 3 structural and 7 non-structural proteins.
One of the characteristic features of positive strand RNA viruses is the interaction of proteins that are involved with viral replication with cellular membranes thus leading to the formation of viral replication centers (RC) in infected cells as discussed in extensio before for Coronavirus’ and Chikungunya Virus (CHIKV). In general, the viral RC contain not viral proteins such as the RNA dependent RNA Polymerase and viral helicase but also dsRNA, thus shielding viral RNA from being recognized by components of the cellular antiviral signalling pathway that recognize viral dsRNA such as RIG-1 and MDA-5 as well as providing a scaffold for the assembly for viral particles. In the case of DENV, WNV, and TBEV, Electron Tomography (ET) studies revealed the presence of different virus induced structures derived from the ER such as cytoplasmic vesicles containing viral proteins and dsRNA that appear to be derived from invaginations of the ER as well as vesicular structures that contain viral proteins within the ER lumen that appear as invaginations of the ER bearing pore-like connections to the cytoplasm as indicated by the co-localisation of either DENV NS1 and NS5 protein or TBEV NS1 protein as well as dsRNA with the ER marker Protein Disulfide Isomerase-1 (PDI), indicating that assembly of the viral RC does take place at the ER but not within the ER of infected cells or BHK-21 cells transfected with a TBEV replicon respectively. As indicated by specific labeling of newly synthesized viral RNA using a MS2 tag, RNA synthesis takes place within ER derived vesicles that are ER invaginations which are connected to the cytoplasm via pore forming channels which – in WNV infected C6/36 cells- are positive for NS3 and NS5. Nascent viral RNA is probably transferred from the sites of viral RNA synthesis to the assembly site by the viral C protein. It should be noted that vesicular structures in TBEV infected BHK-21 cells do not stain positive for dsRNA, NS3 or NS5 proteins but for the viral E and NS1 protein indicating that these vesicular structures might indeed either fuse with viral RNA containing vesicles or that viral RNA is transferred.
Like other mosquitoe borne Flavivirus’, the infection of mosquitoe salivary gland lineage cells (C6/36) cells that are derived from Aedes Albopictus as well as human neuroblastoma (SK-N-SH) cells with ZIKV Paraiba 2019 induce a rearrangement and expansion of the ER with extensive localisation of the viral E protein throughout the cytoplasm and the ER at 72 hrs p.i. with dsRNA present within the ER, indicating that similar to WNV MRC MRM61C infected BHK-21 and DENV-2 infected C6/36 cells, ZIKV RNA is synthesized at the nuclear membrane and within ER invaginations both in C6/36 and SK-N-SH cells with virions intermixed with dsRNA positive vesicles thus indicating that both viral RNA synthesis and assembly of virions are taking place in close proximity during ZIKV replication.
Interestingly, the expansion of the ER, the levels of the viral E protein, the amount of dsRNA staining as well as viral titres despite being similar in pattern and amount in in C6/36 and SK-N-SH cells are delayed by 24 hrs in C6/36. One factor might be that in C6/36 cells ZIKV proteins might inhibit antiviral signalling more efficiently than in mammalian cells or that viral entry in C6/36 cells differs from mammalian thus leading to an activation of antiviral signalling pathways that delay viral replication. Interestingly, CCL-125 cells that are derived from Aedes Agypti do not support the replication of ZIKV Paraiba 2019, suggesting that indeed either viral entry or viral replication is restricted by host specific factors although female Aedes Agypti mosquitoes support the replication of ZIKV BRPE243/2019 and ZIKV /SPH/2019.
In accordance with results obtained in from primary human neuronal cells and from foetal mice brains, the infection of both SK-N-SH and C6/36 with ZIKV Paraiba 2019 induces caspase-3 dependent apoptosis as early as 48 hrs pi. which is preceded by the release of cytochrome-c and mitochondrial depolarisation. In contrast to SK-N-SH cells, C6/36 cells only exhibit a small percentage of cells undergoing apoptosis as indicated by the absence of TUNEL positive cells, indicating that ZIKV might persist in these cells. Considering that the infection of CCL-115 cells with ZIKV Paraiba induces low levels of viral titres, the inhibition of ZIKV replication in CCL-115 cells whilst not complete might prevent apoptosis (maybe by inducing mitophagy of damaged mitochondria) of infected cells and thus allow the transfer of viral particles to humans.
Following the infection with either DENV-2, Semliki Forest Virus (SFV), Bunyamwera Virus or Chikungunya Virus (CHIKV), mosquitoe salivary gland cells produce cytokines that promote the migration of monocyte derived macrophages not only in infected mosquitoes but also following a blood meal in hosts, thus facilitating the infection of those cells, suggesting that despite low viral titres the amount of viral particles might be sufficient to infect human cells, aided by cytokines.
Role of p53 in activating ZIKV induced apoptosis
In ZIKV Paraiba 2019 infected C6/36 and SK-N-SH cells the ER is enlarged indicating ER stress, although it is not clear if the enlargement is dependent on the induction of the UPR by viral proteins such as the viral E, NS4a or prM proteins (similar to DENV) resulting in the formation of autophagosomes (as observed in Varicella Zoster Virus (VZV) infected cells) or not.
As discussed before in extensio, the expression of viral proteins derived from a variety of positive strand RNA viruses including CHIKV and Japanese Encephalitis Virus (JEV) induces the ER stress response that can result in increased formation of autophagosomes in infected cells in a Beclin-1 dependent manner as well as to apoptosis in a p53 independent manner.
Furthermore, ER stress promotes the expression of the p53 isoform p53/47 thus inducing a G2 arrest by preventing the expression of p21 in HCT-116 cells, leading to a stabilisation of 14-3-3σ. Indeed, the infection of human Neural Progenitor Cells (hNPC) with ZIKV MR 766 results in a G2 arrest, suggesting that ZIKV induced ER stress induces a G2 arrest. p53 can however also be activated in ZIKV infected cells following the induction of genotoxic stress as a result of stalled replication forks. In this model, the expression of viral proteins induces the downregulation of genes associated with the repair of damaged DNA as well as of genes associated with DNA replication as discussed in a previous post and which has been demonstrated for ZIKV MR 766 infected hNPC and foetal brains infected with ZIKV SZ01. In ZIKV H/PF/2019 infected hNPC and human i90c16 (induced pluripotent human neural stem cell that are derived from IMR-90 human lung fibroblast cells) (nuclear) Ser-15 phosphorylated p53 and active Caspase-3 can be detected as early as 24 hrs p.i. suggesting that ZIKV indeed does induce the activation of p53 in neural cells. If however p53 is also induced in mosquitoe cells and if the activation of p53 also induces DNA damage repair response or if the DDR is abrogated (and induces apoptosis) has not been determined. ZIKV H/PF/2019 infected i90c16 cells do however display an increase in γH2AX (H2AX-Ser19) positive foci indicative of replicative stress as well as indicating failure to induce the repair of ds/ss DNA breaks, which is supported by findings that mouse foetal brains infected with ZIKV Mex 1-144 exhibit a decrease in Ki67 positive cells, a decrease of Histone H3-P (Ser-10) positive cells as well as a decrease in CldU positive cells with an increased cell cycle length while the percentage of TUNEL positive cells increases slightly, suggesting that ZIKV Mex 1-144 and ZIKV H/PF/2019 induce replicative stress which induces apoptosis in a subset of infected neural cells in the developing foetal brain whilst inhibiting the progression of the cell cycle including mitotic entry. It should be emphasised however that majority of infected neural cells might not undergo apoptosis as indicated by observations that the infection of foetal mice at embryonic day (E) 17.5 with ZIKV Mex 1-144 the majority of infected cells is TUNEL negative; these results are consistent with previous observations that ZIKV associated intrauterine growth restriction is more pronounced in foetal mice that were infected at E 14.5 or earlier.

In addition to genotoxic stress, ZIKV might activate p53 by sequestering Mdm2/HDM2 to the nucleolus similar to WNV Capsid protein since ZIKV antigen(s) have been described to localise to structures tentatively identified as the nucleolus and/or nuclear speckles in infected Vero cells and the DENV Capsid protein co-localises with DAXX in the nucleus, an interaction that has been linked to the induction of apoptosis by DENV C protein . In this scenario, the expression of the viral Capsid protein promotes the stabilisation of p53 by sequestering Mdm2 independent of p19Arf (as well as by binding inhibitor of the serine/threonine phosphatase PP2A (I(2)(PP2A)), thus inducing the expression of pro-apoptotic genes including Bax and subsequently promoting apoptosis via the mitochondrial pathway. In addition, the cellular nucleolar helicase DDX56 relocalises in WNV infected cells from the nucleolus to sites of viral replication, indicating that the infection of cells with WNV does indeed cause nucleolar stress and thus activation of p53.
The question remains however if in addition to genotoxic stress and the nucleolar localisation of the viral Capsid protein the induction of ER stress by the formation of viral RC contributes to the activation of p53 or if the expression of the viral NS4A/NS4B proteins promote the degradation of the cellular Filamin A (FLNA) protein by increasing the nuclear levels of p62/SQSTM-1. In the latter scenario, the inhibition of ER stress induced autophagy by NS4A/NS4B via the inhibition of PI-3K prevents the degradation of p62/SQSTM-1 by selective autophagy, facilitating the proteasomal degradation of nuclear FLNA and thus prevent the recruitment of Rad51 to sites of DNA damage and subsequent activation of the ATR dependent pathway of DNA repair whilst promoting the initiation of (error prone) non-homologues end joining (NHEJ) dependent DNA repair. Promoting NHEJ in the absence of apoptosis therefore might lead to the accumulation of mutations that resemble those observed in genetic cases of microcephaly as described in a recent study that compared data from ZIKV H/PF/2019 infected human NPC with three different mouse models of microcephaly.
 |
| Figure: ZIKV and p62/SQSTM-1 dependent selective autophagy |
The induction of ER stress by viral proteins such as NS4A, NS4B, E or prM or the formation of viral RC therefore might induce apoptosis either in a p53 dependent or independent pathway (via CHOP), promote the proteasomal degradation of FLNA (via p62/SQSTM-1) and might prevent DNA repair by facilitating the translocation of p53 into the cytoplasm by Glycogen Synthase Kinase-3 β (GSK3β) dependent phosphorylation of p53 at Ser-376. In contrast, the localisation of the viral Capsid protein to the nucleus/nucleolus might activate p53 by sequestering Mdm2/HDM2 into the nucleolus and/or nuclear speckles, thus promoting the initiation of the DNA damage response (which in the presence of other viral proteins might be inhibited downstream). In ZIKV infected cells these processes might be present at different times post infection and/or dependent on the cell line/ cell type.
ZIKV and G1 cell cycle arrest: inhibition of CDC-7?
The induction of p53 by ZIKV following the induction of the ER stress response may not only induce a G2 arrest but also delay the progression from G1 to S phase which is regulated by two kinases, CDK-2 and CDC-7. As discussed previously, the infection of foetal mouse brains with ZIKV SZ 01 decreases the expression of genes related to the initiation and progression of DNA replication and the infection of hNPC with ZIKV MR 766 decreases the expression of CDK-2, suggesting that ZIKV infection can indeed at least delay the onset and certainly the progression of S phase which –as described above- is in accordance with recent results obtained from ZIKV Mex 1-14 infected mouse brains.
CDC-7 is the catalytic subunit of the Dbf4b dependent Ser/Thr kinase whose expression is inhibited by p53 via p53 dependent upregulation of miR-192/215 as well as the E3 ubiquitin ligase Fbxw7β following the treatment of IMR90 fibroblast cells with Doxorubicin, low doses of Actinomycin D or γ-irradiation. The activation of p53 in ZIKV infected cells therefore might delay the progression of G1 to S phase by at least two mechanisms; first by downregulating the expression of CDK-2 and second by inhibiting CDC-7 via the expression of miR-192/195 and Fbxw7β.
The DENV Capsid protein not only localises to the nucleus, but also co-localises with Death Associated Protein-6/DAXX in PML nuclear bodies and induces the expression of CD95/ TNFRSF6/Fas, thus inducing apoptosis of HepG2 cells expressing wt DENV Capsid protein but not DENV Capsid protein variants that do not localise to the nucleus due to K73A/K74A or R85A/K86A mutations via the induction of Fas dependent activation of Caspase-3 and -9 following treatment with an anti-Fas/CD95 antibody.
It is therefore possible that the infection of neuronal and non-neuronal cells with ZIKV may trigger the expression of CD95 at the cell surface thus sensitizing infected cells to apoptosis induced by the activation of CD95/FasR either in an autocrine manner and/or by FasL expressing activated T-lymphocytes.
 |
| Figure: ZIKV Capsid protein: Relocalising DAXX to nuclear bodies? |
In summary, ZIKV may induce the activation of p53 by various pathways, including the induction of genotoxic stress as a result of stalled replication forks, the induction of nucleolar stress by relocalising or sequestering nucleolar proteins and the activation of the ER stress response as a result of the formation of viral replication centers, thus resulting in extending the length of the cell cycle by prolonging S phase and delaying the progression of G1 to S phase followed by an arrest in G2 phase of the cell cycle without progressing to Prophase. The localisation of the viral Capsid protein, similar to the Capsid protein of WNV and DENV, might contribute to the activation of intrinsic and extrinsic apoptotic pathways by not only redistributing nucleolar proteins such as Mdm2/HDM2 but also by sequestering DAXX in PML bodies respectively thus sensitizing infected cells to CD95L induced apoptosis. Further research however is needed to elucidate the contribution of individual viral proteins to these processes and the involvement of additional cellular proteins. One possibility is that ZIKV infection prevents the degradation of FAP-1 by inhibiting p62/SQSTM-1 dependent selective autophagy of FAP-1 thus preventing Fas induced apoptosis even in the presence of FasL whereas late in infection intrinsic apoptosis is activated by NS4B as well as the ER stress response and p53. The expression of the viral NS4A and NS4B might not only inhibit PI3K mediated regulation of autophagy but also promoting PI3K mediated apoptosis induced by p53 by preventing Mdm2 mediated degradation of p53, thus enhancing the effects of Mdm2 sequestration by the viral Capsid protein. Further experiments are however warranted to confirm this hypothesis.
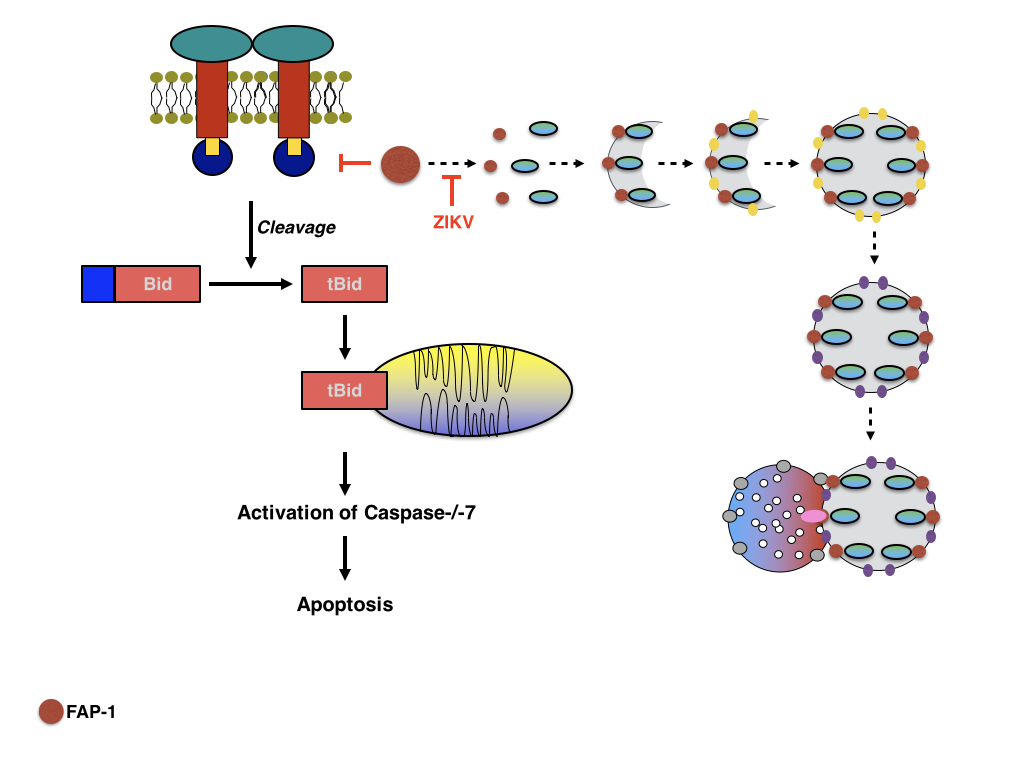 |
| Figure: Inhibition of Fas dependent apoptosis by stabilising Fap-1? |
In addition to investigating the individual contribution of viral proteins to ZIKV induced activation of p53 and ZIKV induced apoptosis, differences between mosquitoe and human derived cell lines have to be examined as well since the infection of CCL-125 cells with ZIKV Paraiba 2019 does not produce high viral titres and infected cells do not undergo viral induced apoptosis, suggesting that either p53 is not activated in CCL-125 cells or that anti-apoptotic pathways are activated.
 |
| Figure: ZIKV and the induction of p53 |
Further reading
Li H, Saucedo-Cuevas L, Shresta S, & Gleeson JG (2019). The Neurobiology of Zika Virus. Neuron, 92 (5), 949-958 PMID: 27930910
Vasilakis N, & Weaver SC (2019). Flavivirus transmission focusing on Zika. Current opinion in virology, 22, 30-35 PMID: 27936448
Offerdahl DK, Dorward DW, Hansen BT, & Bloom ME (2019). Cytoarchitecture of Zika virus infection in human neuroblastoma and Aedes albopictus cell lines. Virology, 501, 54-62 PMID: 27863275
Barreto-Vieira DF, Barth OM, Silva MA, Santos CC, Santos Ada S, F JB Filho, & Filippis AM (2019). Ultrastructure of Zika virus particles in cell cultures. Memorias do Instituto Oswaldo Cruz, 111 (8), 532-4 PMID: 27581122
Junjhon J, Pennington JG, Edwards TJ, Perera R, Lanman J, & Kuhn RJ (2019). Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. Journal of virology, 88 (9), 4687-97 PMID: 24522909
Chatel-Chaix L, & Bartenschlager R (2019). Dengue virus- and hepatitis C virus-induced replication and assembly compartments: the enemy inside--caught in the web. Journal of virology, 88 (11), 5907-11 PMID: 24623440
Gillespie, L., Hoenen, A., Morgan, G., & Mackenzie, J. (2010). The Endoplasmic Reticulum Provides the Membrane Platform for Biogenesis of the Flavivirus Replication Complex Journal of Virology, 84 (20), 10438-10447 DOI: 10.1128/JVI.00986-10
Miorin L, Romero-Brey I, Maiuri P, Hoppe S, Krijnse-Locker J, Bartenschlager R, & Marcello A (2019). Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. Journal of virology, 87 (11), 6469-81 PMID: 23552408
Li H, Saucedo-Cuevas L, Regla-Nava JA, Chai G, Sheets N, Tang W, Terskikh AV, Shresta S, & Gleeson JG (2019). Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell stem cell, 19 (5), 593-598 PMID: 27545505
Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, Ge J, Wang S, Goldman SA, Zlokovic BV, Zhao Z, & Jung JU (2019). Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell stem cell, 19 (5), 663-671 PMID: 27524440
Schuck S, Prinz WA, Thorn KS, Voss C, & Walter P (2009). Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. The Journal of cell biology, 187 (4), 525-36 PMID: 19948500
Ghouzzi VE, Bianchi FT, Molineris I, Mounce BC, Berto GE, Rak M, Lebon S, Aubry L, Tocco C, Gai M, Chiotto AM, Sgrò F, Pallavicini G, Simon-Loriere E, Passemard S, Vignuzzi M, Gressens P, & Di Cunto F (2019). ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly and p53. Cell death & disease, 7 (10) PMID: 27787521
Shao Q, Herrlinger S, Yang SL, Lai F, Moore JM, Brindley MA, & Chen JF (2019). Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development (Cambridge, England), 143 (22), 4127-4136 PMID: 27729407
Wu KY, Zuo GL, Li XF, Ye Q, Deng YQ, Huang XY, Cao WC, Qin CF, & Luo ZG (2019). Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell research, 26 (6), 645-54 PMID: 27174054
Pingen M, Bryden SR, Pondeville E, Schnettler E, Kohl A, Merits A, Fazakerley JK, Graham GJ, & McKimmie CS (2019). Host Inflammatory Response to Mosquito Bites Enhances the Severity of Arbovirus Infection. Immunity, 44 (6), 1455-69 PMID: 27332734
Agarwal A, Joshi G, Nagar DP, Sharma AK, Sukumaran D, Pant SC, Parida MM, & Dash PK (2019). Mosquito saliva induced cutaneous events augment Chikungunya virus replication and disease progression. Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases, 40, 126-35 PMID: 26925703
Hewitt G, Carroll B, Sarallah R, Correia-Melo C, Ogrodnik M, Nelson G, Otten EG, Manni D, Antrobus R, Morgan BA, von Zglinicki T, Jurk D, Seluanov A, Gorbunova V, Johansen T, Passos JF, & Korolchuk VI (2019). SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy, 12 (10), 1917-1930 PMID: 27391408
Yang MR, Lee SR, Oh W, Lee EW, Yeh JY, Nah JJ, Joo YS, Shin J, Lee HW, Pyo S, & Song J (2008). West Nile virus capsid protein induces p53-mediated apoptosis via the sequestration of HDM2 to the nucleolus. Cellular microbiology, 10 (1), 165-76 PMID: 17697133
Reid CR, & Hobman TC (2019). The nucleolar helicase DDX56 redistributes to West Nile virus assembly sites. Virology, 500, 169-177 PMID: 27821284
Balinsky CA, Schmeisser H, Ganesan S, Singh K, Pierson TC, & Zoon KC (2019). Nucleolin interacts with the dengue virus capsid protein and plays a role in formation of infectious virus particles. Journal of virology, 87 (24), 13094-106 PMID: 24027323
Danthi P (2019). Viruses and the Diversity of Cell Death. Annual review of virology, 3 (1), 533-553 PMID: 27501259
Parquet MC, Kumatori A, Hasebe F, Morita K, & Igarashi A (2001). West Nile virus-induced bax-dependent apoptosis. FEBS letters, 500 (1-2), 17-24 PMID: 11434919
Chu JJ, & Ng ML (2003). The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. The Journal of general virology, 84 (Pt 12), 3305-14 PMID: 14645911
Lee CJ, Liao CL, & Lin YL (2005). Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. Journal of virology, 79 (13), 8388-99 PMID: 15956583
Zhao P, Han T, Guo JJ, Zhu SL, Wang J, Ao F, Jing MZ, She YL, Wu ZH, & Ye LB (2012). HCV NS4B induces apoptosis through the mitochondrial death pathway. Virus research, 169 (1), 1-7 PMID: 22542667
Hunt TA, Urbanowski MD, Kakani K, Law LM, Brinton MA, & Hobman TC (2007). Interactions between the West Nile virus capsid protein and the host cell-encoded phosphatase inhibitor, I2PP2A. Cellular microbiology, 9 (11), 2756-66 PMID: 17868381
Buckley A, & Gould EA (1988). Detection of virus-specific antigen in the nuclei or nucleoli of cells infected with Zika or Langat virus. The Journal of general virology, 69 ( Pt 8), 1913-20 PMID: 2841406
Netsawang J, Noisakran S, Puttikhunt C, Kasinrerk W, Wongwiwat W, Malasit P, Yenchitsomanus PT, & Limjindaporn T (2010). Nuclear localization of dengue virus capsid protein is required for DAXX interaction and apoptosis. Virus research, 147 (2), 275-83 PMID: 19944121
Giovannoni F, Damonte EB, & García CC (2019). Cellular promyelocytic leukemia protein is an important dengue virus restriction factor. PloS one, 10 (5) PMID: 25962098
Giovannoni F, Damonte EB, & García CC (2019). Correction: Cellular Promyelocytic Leukemia Protein Is an Important Dengue Virus Restriction Factor. PloS one, 10 (8) PMID: 26313937
Licon Luna RM, Lee E, Müllbacher A, Blanden RV, Langman R, & Lobigs M (2002). Lack of both Fas ligand and perforin protects from flavivirus-mediated encephalitis in mice. Journal of virology, 76 (7), 3202-11 PMID: 11884544
Tudzarova S, Mulholland P, Dey A, Stoeber K, Okorokov AL, & Williams GH (2019). p53 controls CDC7 levels to reinforce G1 cell cycle arrest upon genotoxic stress. Cell cycle (Georgetown, Tex.), 15 (21), 2958-2972 PMID: 27611229
Fang D, Cao Q, & Lou H (2019). Sld3-MCM Interaction Facilitated by Dbf4-Dependent Kinase Defines an Essential Step in Eukaryotic DNA Replication Initiation. Frontiers in microbiology, 7 PMID: 27375603
Waring, P., & Mullbacher, A. (1999). Cell death induced by the Fas/Fas ligand pathway and its role in pathology Immunology and Cell Biology, 77 (4), 312-317 DOI: 10.1046/j.1440-1711.1999.00837.x
Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, & Thorburn A (2019). Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nature cell biology, 16 (1), 47-54 PMID: 24316673
Park JS, Oh SY, Lee DH, Lee YS, Sung SH, Ji HW, Lee MJ, Lee YH, Rhee SG, & Bae SH (2019). p62/SQSTM1 is required for the protection against endoplasmic reticulum stress-induced apoptotic cell death. Free radical research, 50 (12), 1408-1421 PMID: 27780373
Urbanowski MD, & Hobman TC (2019). The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism. Journal of virology, 87 (2), 872-81 PMID: 23115297
Abraham AG, & O'Neill E (2019). PI3K/Akt-mediated regulation of p53 in cancer. Biochemical Society transactions, 42 (4), 798-803 PMID: 25109960
No comments:
Post a Comment